
UROP reveal!! by Aiden H. '28
it's like a gender reveal but important
It was inevitable. My MIT prophecy has been fulfilled. There was no path to nor desire for avoidance. I’ve started doing research.01 At apparently one of the worst times in American history to do so. Mind you, how could I not? Over 90% of MIT students will UROP02 Undergraduate Research Opportunities Program, aka undergraduate research, also what this happens to be about. at some point, and January is just too dark and depressing for me not to have something to stress over. And as much as I have kept my blog-persona STEM-free and relatively non-nerdy in order to preserve the major amounts of street cred I have, I fear it must be discussed.
Late last semester, I sent in a bunch of UROP applications on ELx03 The website where ELOs (Experiential Learning Opportunities) are posted, including UROPs. and cold-emailed some labs to try and have a UROP for IAP04 Independent Activities Period - the month of January where students can conduct 'independent activities' before the spring semester starts in February. and spring, and wasn’t even met with rejection, just plain non-response.05 No, I do not subscribe to the mentality that just because you are an adult or busy or both that you cannot respond to emails in your inbox when I know for a fact you check your email 785497293471 times a day, especially considering if a student didn't respond to an authority/higher-up we would be ruined. One PhD student did respond though, we met a couple times before winter break to discuss what a project could look like, and now we meet once a week while I show him all of my data and explains to me what it means and what I need to do with it.
So, for the past month (and the rest of the semester), all of my non-class related energy is being put to my first UROP.
Without further ado, my project is…


As a prospective 6-706 An interdisciplinary major for computer science (6) and molecular biology (7). (or 10-B,07 Chemical-Biological Engineering. Different from 20 (Biological Engineering). we’ll see), this project is actually very relevant and pretty much what a lot of computational biologists are spending they’re time doing doing right now, which is cool because I feel important, but scary because I feel stupid compared to the other PhD people doing this.
From what I’ve heard (although it just generally varies greatly from project to project), this is a fairly unique UROP experience/project/dyanmic? And I’m loving it. While a lot of CS and/or bio UROPs are a part of a much larger project and a group of PhD students, often with multiple UROP students per project or group of projects, mine is just this one PhD student walking me through a short project from beginning to end on whatever we found interesting. Instead of having to acclimate to a lab that has been running for years, I got to go through data selection and project design individually with my mentor, and I’m the only person responsible for actually doing any of the lab (running the code, making the graphs, etc. etc.). This last part is especially freeing because I’m not rushing to match other people’s deadlines, so if I’m slammed one week there’s not any major repercussions.
With the lore and details out of the way, it’s time to get serious.
⚠️⚠️ WARNING: MILDLY INTENSE SCIENCE AHEAD08 I am preemptively sorry to my advisor for any and all mistakes I make in explaining this. ⚠️⚠️
The crux of the project is something that is pretty common throughout comp bio right now as a result of developments towards free-to-use AI for microbiology. Basically, why wait millennia for nature to spurt out better proteins when I could try thousands of possibilities on my computer right now? I won’t go too much into the background of this issue or how it’s currently being solved, but this Veritasium video that was just posted explains everything very well if you’re interested or need a bio refresher.
The project is being mentally split into three stages:
- Data Selection/Collection – Choose a common enzyme family as a starting point, randomly sample the amino acid sequences of a bunch of its variants, and predict their structures computationally. Notably, this meaning finding a way to numerically describe the shape and “fitness” of the active site so that active sites can be compared as better/worse later on.
- Mutate, mutate, mutate – Using existing structures, computationally “mutate” and “evolve” the sampled proteins sequences, and compare the structures of those mutated amino acid sequences to that of their originals (directed evolution). This kinda simulates evolution by just randomly mixing up sequences and seeing if anything better comes of it, but instead of having to have things die, I just press delete!
- Develop a model – Train a model (genetic algorithm) using the results from part 2 to understand how different mutations directly change the shape of active sites. Once the model is trained enough, it should be able to work both ways. Given a sequence, it should find the part of the sequence that describes the active site and be able to mutate it for the better. Given a desired active site, it should be able to back out the amino acid sequence that would result in said structure.
So far, I’m working on parts 1 and 2, with 1 mostly done.
My advisor and I09 Meaning mostly my advisor because he has a lot more knowledge/wisdom/expertise. decided on starting with studying Cytochrome P450, a gigantic protein family that is easily accessible, large enough to sample, and well studied (as well as, like, biologically important but aren’t kinda most enzymes?).
I started by using UniProt, an online protein database, and searched the Cytochrome P450 family. The data in UniProt is organized hierarchically; family –> cluster –> sequence. Searching the P450 family lists clusters, which are just groups of sequences, big or small, that are found to be similar enough on an amino acid level, but all individually create one of the proteins in the Cytochrome P450 family. Cytochrome P450 has >1,000,000 different clusters, so I used UniRef50 (a program/search tool inside UniProt) that simplifies the sequences based on similarity. With a similarity of 90% or 100% (UniRef90 or UniRef100), sequences will be counted if they are 90% or 100% unique, respectively, leaving more sequences. UniRef50 only leaves sequences that are at least 50% unique, and I make the assumption that sequences that are over 50% similar will have nearly identical structures in the end. This is a typical time vs accuracy tradeoff (I as one person cannot run 1,000,000 sequences, but running that many would technically be more accurate in the end).
This leaves >90,000 clusters. From here, I downloaded sequences from the top 10 clusters with the most sequences. Amino acid sequences are downloaded as .fasta files, which are formatted with a “>” followed by the sequence name, and then the amino acid sequence on the following line.

Example fasta file (with only one sequence)
From there I randomly sampled 100 sequences from each cluster file, leaving me with 1000 sequences across 10 clusters that, after all the parsing, should be representative of the sequence population of >1,000,000.
Side note: You’ll save a bunch of time if you remember to check that there are no repeat sequences after you sample and not after you have a bunch of data already and it’s like 1:00 A.M. and you’re pissed! 🎉🎉

what doesn’t kill you makes you stronger – Kelly Clarkson
After this, I downloaded localcolabfold to run AlphaFold 2 (AF, henceforth), an AI system created by Google’s DeepMind that famously solved Levinthal’s paradox.10 The idea that humans could never accurately predict nor randomly guess how proteins fold because the sheer number of proteins and the length of amino acid sequences is way too much data for the human brain to process. Levinthal is also an MIT alum. Since my MacBook doesn’t have the computing capacity to carry out running an AI system of this scale to any capacity, I secure shell log in to a GPU provided by MIT. Every time I ssh in, it gives me a cute picture:



Once I have access to the GPU, I run AF and get met with what can only be described as a shit ton of stuff.

running AF on three separate GPUs because I’m impatient
I had AF set to run 5 “recycles” of each sequence, meaning that each sequence is run 5 times, each prediction theoretically getting stronger (as it has the information of the previous cycles). Most importantly, I get a pLDDT (predicted local distance difference test) score for each cycle the sequence is run. This higher the pLDDT score (>80, mostly), the more accurate the prediction, and the AF output can be assumed to be nearly identical to what the actual protein would look like.
📣📣 Don’t underestimate or under-appreciate this! A computer was literally just given a list of capital letters and it’s accurately predicting how the basis of most microbiology folds and forms!! 📣📣
These pLDDT scores also give me a bunch of other files, most of which aren’t desperately important for me to explain. One fun one, though, is .pdb files, which give me the structure prediction. Using a separate software (Visual Molecular Dynamics), I can upload the file and mess around the with shape. When I get bored I treat it like MS Paint but for biomolecules.

brat but it’s a protein but definitely still brat because it keeps us alive
Another important output is the .json files, which pair sequence-pLDDT as keys-values in a dictionary. This allows me to run the next part, Evolutionary Scale Modeling (ESM), a huge11 >1 trillion teraflops of power and a dataset of >2.5 billion proteins. model made by ex-Meta scientists. This is a transformer-based language model that allows me to turn the sequences into numerical data, vectorize them all into a huge array, and start to look at the differences between each of the clusters. This also gives me cute, encouraging messages.


Eventually12 aka once I stop procrastinating by writing about it on this blog I will graph all of these to visualize exactly how representative my samples are of the population (color the original P450 and the cluster sequences differently, and see if I can see any of the original color). I will also start running the “mutation” part of the plan in the coming weeks, running an already created generative algorithm that mutates by essentially “swapping” amino acid sequences. I will choose a specific part of the sequence that two sequences share, and have everything before that point on the sequences stay as their original, and the rest switch. Since I’m trying to hone the active sites of the enzymes, I will have to find markers in the sequences that represent the amino acids that describe the shape of the active site, and chose this as the point for ESM to run. I drew a picture to explain because words are hard.
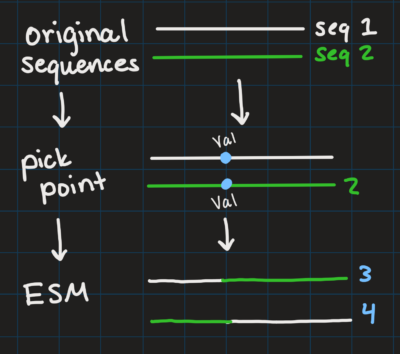
The output is a bunch of new sequences that are “randomly” evolved (except not because I’m making sure it’s evolving the active site). Most of them will probably be awful, but done enough times, I will get a new sequence that has an even better active site than either of the original sequences fed in, and BAM! every world issue is solved.
I’ll use all this in months to start developing a model, but I haven’t gotten that far so I am in no way qualified to even to try and talk about it.
Maybe I’ll make an update when I get that far, but for now, this is my life! Hopeful this inspired someone to do 6-7 and didn’t incite fear into anyone.
- At apparently one of the worst times in American history to do so. back to text ↑
- Undergraduate Research Opportunities Program, aka undergraduate research, also what this happens to be about. back to text ↑
- The website where ELOs (Experiential Learning Opportunities) are posted, including UROPs. back to text ↑
- Independent Activities Period - the month of January where students can conduct 'independent activities' before the spring semester starts in February. back to text ↑
- No, I do not subscribe to the mentality that just because you are an adult or busy or both that you cannot respond to emails in your inbox when I know for a fact you check your email 785497293471 times a day, especially considering if a student didn't respond to an authority/higher-up we would be ruined. back to text ↑
- An interdisciplinary major for computer science (6) and molecular biology (7). back to text ↑
- Chemical-Biological Engineering. Different from 20 (Biological Engineering). back to text ↑
- I am preemptively sorry to my advisor for any and all mistakes I make in explaining this. back to text ↑
- Meaning mostly my advisor because he has a lot more knowledge/wisdom/expertise. back to text ↑
- The idea that humans could never accurately predict nor randomly guess how proteins fold because the sheer number of proteins and the length of amino acid sequences is way too much data for the human brain to process. Levinthal is also an MIT alum. back to text ↑
- >1 trillion teraflops of power and a dataset of >2.5 billion proteins. back to text ↑
- aka once I stop procrastinating by writing about it on this blog back to text ↑